| Marchena Rodríguez, Leticia. Doctora en Odontología. Universidad de Sevilla. Fernández Ortega, Carlos Mª. Odontólogo. Máster en Salud Pública Oral. Universidad de Sevilla.
RESUMEN
Introducción: La Dentinogénesis Imperfecta (DI) es una alteración hereditaria que se origina en la etapa de histodiferenciación durante la odontogénesis. La DI se ha descrito como una alteración hereditaria de carácter autosómico dominante, que se origina durante la etapa de histodiferenciación en el desarrollo dental; constituye una forma de displasia mesodérmica localizada y caracterizada por una expresa alteración de las proteínas de la dentina. La incidencia se estima alrededor de 1/8.000. La DI se divide a su vez en tres grupos: DI tipo I, DI tipo II o Capdepont y DI tipo III o Brandywine.
Objetivos: Conocer las características clínicas, radiográficas e histológicas de los dientes afectados con la DI, para un mejor diagnóstico y tratamiento de estos pacientes.
Material y Métodos: Se ha hecho una revisión bibliográfica en PubMed, Scopus; insertando palabras clave como "Dentinogénesis Imperfecta, Osteogénesis Imperfecta, dentina, esmalte, morfología, genética, diagnóstico y tratamiento" en los últimos años.
Resultados: Paciente de 16 años de sexo masculino que acude a la consulta para una revisión. A nivel médico, el paciente refiere que presenta Osteogénesis imperfecta con fracturas múltiples de varios huesos, como fémur, peroné, húmero hace unos años.
Discusión: Existe un consenso general para el diagnóstico de la Dentinogénesis Imperfecta, el cual se basa fundamentalmente en la genética, las características clínicas e imagenológicas y los estudios genéticos moleculares. Según investigadores, han asociado mutaciones en la DI tipo I-II-III; en concreto en el gen que codifica la dentina sialofosfoproteina (DSPP); uno de los responsables de la biomineralización de la dentina.
Conclusiones: La genética molecular juega un papel fundamental en identificar las causas de la DI en la familia actual. Este caso ilustra la aparición de esta enfermedad en diferentes partes del mundo, personas y etnias.
PALABRAS CLAVE Dentinogénesis Imperfecta, Osteogénesis Imperfecta, dentina, esmalte, morfología, genética, diagnóstico y tratamiento. ABSTRACT Introduction: Dentinogenesis Imperfecta (DI) is a hereditary disorder that originates in histodifferentiation stage during odontogenesis. The DI has been described as an inherited autosomal dominant disorder that arises during the period of tooth development histodifferentiation; It is a form of mesodermal dysplasia characterized by a localized alteration of proteins expressed dentin. The incidence is estimated at around 1 / 8,000. The DI is divided into three groups: DI type I, type II or DI and DI Capdepont type III or Brandywine.
Objectives: To determine the clinical, radiographic and histological affected teeth with DI, for better diagnosis and treatment of these patients characteristics.
Material and Methods: We made a literature review in PubMed, Scopus; inserting keywords like "Dentinogenesis Imperfecta Osteogenesis Imperfecta, dentin, enamel, morphology, genetics, diagnosis and treatment" in recent years.
Results: Patient 16 year old male who comes to the clinic for a review. A medical level, the patient reported having Osteogenesis Imperfecta with multiple fractures of various bones like femur, fibula, humerus few years ago.
Discussion: There is general consensus for the diagnosis of dentinogenesis Imperfecta, which is mainly based on genetic, clinical and imaging features and the molecular genetic studies. Researchers say they have linked mutations in the DI type I-II-III; particularly in the gene encoding sialofosfoproteina dentin (DSPP); one of those responsible for the biomineralization of the dentine.
Conclusions: The molecular genetics play a key role in identifying the causes of the DI in the current family. This case illustrates the emergence of this disease in different parts of the world, people and ethnicities. ABSTRACT The dentinogenesis imperfecta (DI) has been described as an inherited autosomal dominant disorder that arises during the period of tooth development histodifferentiation; is a form of mesodermal dysplasia characterized by a localized alteration of proteins expressed dentin. The incidence is estimated at around 1 / 8,000. The DI is subdivided into three groups: DI type I, type II or Capdepont DI and DI type III or Brandywine.
DI Type I is a genetic disease associated with Osteogenesis Imperfecta (OI), caused by mutations in genes encoding the type I collagen, causing an alteration in the shape of the dentin. The DI type I is a disease that appears always associated with OI. However, only about 10 to 50% of patients suffering from OI have DI. In the DI there is a gray opalescent associated with high attrition affecting both dentitions.
As an alternative treatment at present, the use of composite resins for coronal remodeled, complete with front teeth crowns in the anterior and posterior arises; chrome steel crowns on primary teeth and permanent teeth, wearing overdentures is indicated. KEYWORDS Dentinogenesis Imperfecta Osteogenesis Imperfecta, dentin, enamel, morphology, genetics, diagnosis and treatment. INTRODUCCIÓN
La Dentinogénesis Imperfecta (DI) es una alteración hereditaria que se origina en la etapa de histodiferenciación durante la odontogénesis. La DI se ha descrito como una alteración hereditaria de carácter autosómico dominante, que se origina durante la etapa de histodiferenciación en el desarrollo dental; constituye una forma de displasia mesodérmica localizada y caracterizada por una expresa alteración de las proteínas de la dentina.[1,2] Ésa adopta un aspecto opalescente característico, por lo cual también se denomina Dentina Opalescente Hereditaria. Puede constituir un trastorno primario y exclusivo de la dentina, o formar parte de un síndrome más complejo denominado Osteogénesis Imperfecta (OI).
La incidencia se estima alrededor de 1/8.000. [2,3]
Shields [4] dividió a la Dentinogénesis Imperfecta en tres grupos:
- La Dentinogénesis Imperfecta Tipo I.
- La Dentinogénesis Imperfecta Tipo II ( o Dientes de Capdepont)
- La Dentinogénesis Imperfecta Tipo III (o Brandywine). Es la más rara de aparición. [4,5] La DI tipo I se asocia a la Osteogénesis Imperfecta, sin embargo los tipo II y III aparecen como entidades aisladas. [6]
La DI tipo I es una enfermedad genética, asociada a la Osteogénesis Imperfecta, causada por mutaciones en los genes que codifican el colágeno tipo I, causando una alteración en la forma de la dentina. [7] La DI tipo I es una enfermedad que aparece siempre asociada a la OI. Sin embargo, sólo entre un 10 a un 50% de los pacientes que padecen OI presentan DI.
La dentición temporal se afecta con más severidad que la permanente. Las afectaciones son mayores en los dientes formados en estadíos tempranos. [8] Manifestaciones clínicas de la Dentinogénesis Imperfecta Tipo I Las coronas de los dientes de ambas denticiones presentan un color ámbar translúcido y opalescente. Existen también variabilidades morfológicas como formas grandes, cortas, normales, abrasionadas, con forma de campana o presentando varias de estas características. [9,10]
El esmalte sobre la dentina afectada tiende a "astillarse", por el borde incisal de dientes anteriores; por el surco oclusal de dientes posteriores y por los surcos linguales y bucales de todos los dientes; debido a ello, la dentina queda expuesta y sufre un desgaste muy rápido que puede llegar a ser muy grave. [11]
Radiográficamente, las coronas dentarias son bulbosas, con una constricción marcada a nivel cervical, raíces cortas y delgadas con reducción del tamaño u obliteración de la cámara pulpar que puede afectar también al sistema de conductos radiculares. [12]
Schwartz y Tsiporras comprobaron que la rapidez de la obliteración pulpar puede ser diferente dependiendo del tipo de DI que se padezca, siendo más rápida en la DI tipo III. [13]
A nivel histológico, se presenta dentina anormal, pudiendo existir zonas atubulares, y zonas con túbulos en número reducido y tamaño variable. Lindau [14] observaron que la mayoría de las alteraciones de la dentina se producen cerca del esmalte, por lo que sugieren que estas anomalías dentinarias se originan en estadíos tempranos de la DI.
En el interior de la cámara pulpar es frecuente encontrar cuerpos calcificados o dentículos verdaderos. [14,15]
Manifestaciones clínicas de la Dentinogénesis Imperfecta tipo II
Solo se presentan anormalidades de la dentina sin enfermedad ósea. La severidad del defecto varía con la edad y tipo de órgano dental. La dentición primaria se encuentra severamente afectada; principalmente los incisivos y los primeros molares. [16]
En la dentición permanente, los más afectados son los incisivos y los primeros molares permanentes, seguidos de los premolares, caninos y por último los segundos y terceros molares. En los dientes afectados se encuentra mayor concentración de glucosaminoglicanos. [17,18]
A nivel de la cámara pulpar, la cámara y conductos radiculares son parcial o totalmente obliterados. [19] Manifestaciones clínicas de la Dentinogénesis Imperfecta tipo III o Dentina Opalescente de Brandywine También llamada Dentina Opalescente de Brandywine, Maryland; debido a la gran población de pacientes en esta localidad, con este tipo de trastorno. Es menos severa que el tipo II, semejante a la producida por la ingesta de alcohol y la aplasia del esmalte. Se describe como dientes de cáscara, en los cuales el esmalte aparece normal y la dentina es extremadamente delgada. [20]
Actualmente se está proponiendo una reclasificación de la Dentinogénesis Imperfecta, considerando a los Tipo II y III como una sola entidad, debido a que presentan características similares y la misma alteración genética. Desde el punto de vista clínico, los tres tipos, en ambas denticiones, comparten diversas características: Un aspecto opalescente con una translucidez rara y de color que varía del café amarillo al gris azulado. Este cambio de coloración es debido a la dentina anormal subyacente. Se presentan exposiciones pulpares múltiples y radiolucideces periapicales. [21] Witkop, [22] establece que existe gran atricción, la cual es mayor en los dientes primarios que en los permanentes. La atricción y la eventual pérdida de las coronas pueden causar hiperplasia de las crestas alveolares, lo que puede inducir en su incremento, a la neoformación de hueso alveolar y fibrosis gingival. La dimensión vertical de la cara se pierda, los músculos sin soporte colapsan la expresión facial, el bermellón de los labios desaparece y los pliegues mentolabiales y nasolabiales se profundizan.
Características radiográficas de la Dentinogénesis Imperfecta
Radiográficamente la Dentinogénesis Imperfecta (DI) se caracteriza por la presencia de dientes con coronas bulbosas, constricción en el cuello y raíces cortas y delgadas. Casi siempre se puede observar obliteración precoz, parcial o total, de las cámaras pulpares por la formación continua de dentina. [23] Diagnóstico Diferencial de la Dentinogénesis Imperfecta Es importante establecer el diagnóstico diferencial de la DI con la displasia de la dentina, conocida también como "dientes sin raíz". En la DI existe un color gris opalescente asociado con gran atricción que afecta a ambas denticiones, mientras que la DI tipo II, la atricción y la decoloración está limitada a la dentición temporal y en la DI tipo II, los dientes son clínicamente normales. Tratamiento de la Dentinogénesis Imperfecta Como tratamiento alternativo en la actualidad, se plantea el uso de resinas compuestas para el remodelado coronal, coronas completas con frente estético en los dientes anteriores y en los posteriores; coronas de acero-croma, esto en dentición temporal.
En dentición permanente, la utilización de sobredentaduras junto con la realización de tratamientos de endodoncia, en los casos en los que sea posible la localización de los conductos radiculares.
En relación con las posibilidades del tratamiento protésico o prostodóntico, se debe recordar que la preparación de la DI; la dentina presenta gran atricción por la mala calidad del esmalte; por lo que para la preparación de la corona se tiene que remover todo el tejido afectado. Sin embargo, hay condiciones que favorecen el tratamiento, como la no necesidad de utilizar anestesia para obliteración de los conductos radiculares. Por otro lado, los dientes afectados por la DI presentan baja incidencia de caries y un tejido periodontal sano.
Durante las exodoncias debemos tener un extremo cuidado con el riesgo de producir fracturas mandibulares y fracturas dentarias; éstas últimas debidas a la presión con el fórceps. [24] Además la alteración del tejido conectivo puede producir, en algunos pacientes; vasculopatías y trastornos de la hemostasia y de la cicatrización, con el consiguiente riesgo de hemorragias. [25,26] OBJETIVO GENERAL Conocer las características clínicas, radiográficas e histológicas de los dientes afectados con la DI, para un mejor diagnóstico y tratamiento de estos pacientes. MATERIAL Y MÉTODOS Se ha hecho una revisión bibliográfica en PubMed, Scopus; insertando palabras clave como "Dentinogénesis Imperfecta, Osteogénesis Imperfecta, dentina, esmalte, morfología, genética, diagnóstico y tratamiento" en los últimos 15 años. A propósito del tema desarrollamos un caso clínico de la temática que nos ocupa. RESULTADOS Caso clínico Paciente de 16 años de sexo masculino que acude a la consulta para una revisión. A nivel médico, el paciente refiere que presenta Osteogénesis imperfecta con fracturas múltiples de varios huesos, como fémur, peroné, húmero hace unos años. Ha estado en tratamiento con Hormona del Crecimiento desde los 4 años y posteriormente estuvo en tratamiento con Pamidronato durante 8 meses y viendo la remisión de las fracturas; se decide reducir el tiempo de administración, a 6 meses al año. Su otro hermano de 7 años, también acudió a la consulta para revisión y no presentaba Osteogénesis imperfecta ni afectación dentinaria
Tras la exploración bucodental con espejo y sonda exploratoria, apreciamos mala higiene oral, con acumulo de tártaro y encías insertadas eritematosas e inflamadas en 11,12,13,14,15,21,22,23,24; con sangrado al mínimo contacto.
Presenta caries con serio compromiso pulpar del 16-26-36-46 y se aprecia un ligero cambio de coloración en el sector anterosuperior e inferior; de un tono gris azulado; aunque la morfología coronaria es aparentemente normal y la capa de esmalte no presenta zonas astilladas, ni zonas expuestas de dentina.
Como propuesta de tratamiento se decide restaurar las caries de los molares permanentes superiores e inferiores; colocando cemento de vidrio ionómero y posteriormente restauración con resina compuesta.
Se le realiza tartrectomía y pulido de superficies dentales, junto con la aplicación de flúor e instrucciones de higiene oral. Se le vuelve a dar cita para revisiones cada 6 meses; y comprobar el estado dental y periodontal del paciente; motivándole en el reforzamiento del cepillado y control de hábitos higiénicos-dietéticos. 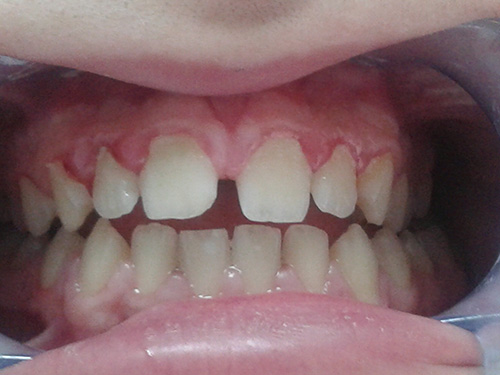 Figura.1: Papilas interdentales inflamadas y eritematosas por falta de cepillado, junto con un cambio de coloración gris azulado en los incisivos superiores e inferiores.
DISCUSIÓN
Existe un consenso general para el diagnóstico de la Dentinogénesis Imperfecta, el cual se basa fundamentalmente en la genética, las características clínicas e imagenológicas y los estudios genéticos moleculares. Trejos y colaboradores [27] definen la DI como una alteración hereditaria que se origina en la etapa de histodiferenciación de la Odontogénesis, constituyendo una forma de displasia mesodérmica localizada, cuya característica especial es una alteración específica de las proteínas de la dentina. Gallusi [28] mostró un caso de DI tipo III con decoloración intensa de Incisivos inferiores en comparación con los Incisivos superiores. A nivel de maloclusión, se presentó mordida cruzada con pérdida de dimensión vertical debido al desgaste de las coronas; exponiéndose la dentina.
MacDougall [29] ha asociado mutaciones en la DI tipo I-II-III; en concreto en el gen que codifica la dentina sialofosfoproteina (DSPP). Este gen codifica dos proteínas principales de la matriz no colágena, la dentina sialoproteína (DSP) y la fosfoproteína de la dentina (también conocida como fosforín) codificados en el cromosoma 4q21.3.
Según Yamahoshi [30] la glicoproteína dentinaria (DGP) se ha propuesto como eje principal en la biomineralización dentinaria.
Kim [31] en 2005 demostraron que dos sujetos sin relación afín, tenían mutaciones idénticas en la secuencia del gen que codifica la dentina sialofosfoproteína (DSPP), en concreto en el nucleótido 1191; se cambia la guanina a timina y el cambio del aminoácido 18 de valina a fenilalanina en el exón 3. Por lo tanto se han propuesto que la dentina opalescente hereditaria tipo II debe ser usada para descubrir tanto la DI tipo II y la tipo III.
Parece ser que esta mutación en el primer nucleótido del exón 3, es un punto de acceso a la aparición de la DI. En cuanto a la dentina fosfoproteína (DPP) se encontraron 12 casos de mutaciones en el exón 5 del gen de la dentina sialofosfoproteína (DSPP); 4 de ellos fueron descritos por Nieminen en 2011, quien investigó los orígenes étnicos. [32]
CONCLUSIÓN
La genética molecular juega un papel fundamental en identificar las causas de la DI en la familia actual. Este caso ilustra la aparición de esta enfermedad en diferentes partes del mundo, personas y etnias.
Es importante un diagnóstico precoz de estas anomalías dentales, ya que el tratamiento dental es difícil y complejo para rehabilitar la función estética y masticatoria.
BIBLIOGRAFÍA
1. Sapir S, Shapira J. Dentinogenesis imperfecta: an early treatment strategy. Pediatr Dent 2001;23:232-7.
2. Cauwels R, De Coster P, Mortier G, Marks L. Dentinogenesis imperfecta associated with short stature, hearing loss and mental retardation: a new síndrome with autosomal recessive inheritance? J Oral Patho Med 2005;34:4446.
3. Rios D, Vieira AL, Tenuta LM, Machado MA. Osteogénesis imperfecta and Dentinogénesis imperfecta: associated disorders. Quintessence Int 2005;36(9):695-701.
4. Shields ED, Bixler D, EI-Kafrawy. A proposed classification for heritable human dentine defects with a description of a new entity. Archs Oral Biol 1973;18:543-53.
5. Kamboj M, Chandra A. Dentinogenesis imperfecta type II: an affected family saga. J Oral Sci 2007;49(3):241-4.
6. Barbería Leache E, Boj Quesada JR, Catalá Pizarro M, García Ballesta C, Mendoza Mendoza A. Odontopediatría. 2ºed. Barcelona: Masson 2001.
7. Malmgren B, Lindskog S. Assesment of dysplastic dentin in osteogenesis imperfecta and dentinogenesis imperfecta. Acat Odontol Scand 2003;61(2):72-80.
8. O`Connell AC, Marini JC. Evaluation of oral problems in an osteogenesis imperfecta population. Oral Surg Oral Med Oral Pathol Radiol Endod. 1999;87:189-96.
9. Malmgren B, Norgren S. Dental aberrations in children and adolescents with osteogenesis imperfecta. Acta Odontol Scand 2002;60(2):65-71.
10. Huber MA. Osteogenesis imperfecta. Oral Surg Oral Med Oral Pathol Radiol Endod 2007;103(3):314-20.
11. Huq N, Loganathan A, Cross K. Association of bovine dentine phosphophoryn with collagen fragments. Oral Biol 2005;50:817-9.
12. Malogren B, Lindskog S, Elgadi A. Clinical, histologic, and genetic investigation in two large familias with dentinogenesis imperfecta type II. Human Gen 2004;114:491-8.
13. Schwartz S, Tsipouras P. Oral findings in osteogenesis imperfecta. Oral Surg Oral Med Oral Pathol 1984;57(2):161-7.
14. Lindau BM, Dietz W, Hoyer I, Lundgren T, Storhaug K, Norén JG. Morphology of dental enamel and dentine-anamel junction in osteogenesis imperfecta. Int J Paediatr Dent 1999;9:13-21.
15. Lindau BM, Dietz W, Hoyer I, Storhaug K, Norén JG. Discriminationn of morphological findings in dentine from osteogenesis imperfecta patients using combinations of polarized light microscopy, microradiography, and scanning electron miscroscopy. Int J Paediatr Dent 1999;9:253-61.
16. Engelbert RH, Uiterwaal CS, Gulmans VA, Pruijs H, Helders PJ. Osteogenesis imperfecta in childhood: prognosis for walking. J Pediatr 2000;137(3):397-402.
17. Sreenath T, Thyagarajan T, May B. Dentin Sialophosphoprotein knockout mouse teeth display widened predentin mineralization similar to human dentinogenesis imperfecta type II. J Biol Chem. 2003;278(27):24874-24880.
18. Sánchez YAE. Tratamiento prostodóntico en paciente con dentinogénesis imperfecta. Reporte de un caso. Acta Odontol Venez. 2000;38:44-50.
19. MacDougall M.Dental structural diseases mapping to human chromosome 4q21. Connect Tissue Res. 2003;44:285-91.
20. Rauch F, Plotkin H, Travers R, Zeitlin L, Glorieux FH. Osteogenesis imperfecta types I, III and IV. Effect of pamidronate therapy on bone and mineral metabolism. J Clin Endocrinol Metab 2003;88(3):986-92.
21. Cordoves M, García I, Suárez A. Revisión bibliográfica sobre la Osteogénesis Imperfecta de las publicaciones más relevantes aparecidas los últimos 12 meses. Universida Rey Juan Carlos. Madrid,España, 2008.
22. Sánchez AE. Tratamieto prostodóntico en pacientes con dentinogénesis imperfeca. Reporte de un caso. Acta Odontológica Venezolana 2010;38(2):49-55.
23. White S, Pharoah M. Principios e interpretación en Radiología Oral, 4ta Ed. Elsevier. Madrid, 2002.
24. Stephen LX, Beighton P. Dental management of severe dentinogenesis imperfecta in a mild form of osteogenesis imperfecta. J Clin Pediatr Dent 2002;26(2):131-6.
25. Bullón P, Machuca G. Tratamiento odontológico en pacientes especiales. Madrid;2004.
26. Prabhu N, Duckmanton N, Stevenson A, Cameron A. The placement of osseointegrated dental implants in a patient with type IV B osteogenesis imperfecta: a 9-year follow-up. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007;103:349-54.
27. Trejos P,Hernando V, De León C. Dentinogénesis Imperfecta: Reporte de un caso. Revista Estomatología 2007;15(2):19-27.
28. Gallusi G, Libonati A, Campanella V. SEM-morphology in dentinogenesis imperfecta type II: microscopic anatomy and efficacy of a dentine bonding system. Eur J Paediatr Dent 2006;7:9-17.
29. MacDougall M, DuPont B, Simmons D, Reus B, Krebsbach P, Karrman C, et al. Ameloblatin gene (AMBN) maps within the critical region for autossomal dominant amelogenesis imperfecta at chromosome 4q21. Genomics 1997;41:115-8.
30. Yamakoshi Y, Hu JC, Fukae M, Zhang H, Simmer JP. Dentin glycoprotein: the protein in the middle of the dentin sialophosphoprotein chimera. J Biol Chem.2005;280:17472-9.
31. Kim JW, Hu JC, Lee JI, Moon SK, Kim YJ, Jang KT, et al. Mutation hot spot in the DSPP gene causing dentinogenesis imperfecta type II. Hum Genet.2005;116:186-91.
32. Nieminen P, Papagiannoulis-Lascarides L, Waltimo-Siren J, Ollila P, Karjalainen S, Arte S, et al. Frameshift mutations in dentin phosphoproteinand dependence of dentin disease phenotype on mutation location. J Bone Miner Res. 2011;26:873-80.
|