| Marchena Rodríguez, Leticia. Odontóloga. Máster en Salud Pública Oral. Universidad de Sevilla. Fernández Ortega, Carlos Mª. Odontólogo. Máster en Salud Pública. Universidad de Sevilla.
RESUMEN
El Síndrome de Q-T largo se define como una anomalía del sistema eléctrico del corazón. Se ha denominado una canalopatía, una enfermedad de canales iónicos. El síndrome de Q-T largo es causado por una mutación en los genes para canales iónicos a nivel cardiaco para el potasio, calcio y sodio, dependiente del cromosoma afectado. Así se determinará si se trata de un síndrome de Romano Ward (5 subtipos) Jervel Lange Nielsen (2 subtipos) y dos subtipos denominados síndrome de Timothy (afecta los canales de calcio) y el síndrome de Anderson (afecta a canales de potasio).
La bradicardia fetal puede ser una de las primeras manifestaciones clínicas en el síndrome de Q-T largo. Si la sospecha de la enfermedad es muy alta, la amniocentesis a partir de las 16 semanas de gestación, puede ser de utilidad para el diagnóstico. Existen ciertos fármacos que pueden prolongar el intervalo QT, en concreto la taquicardia helicoidal secundaria a fármacos no antiarrítmicos es un evento raro; pero se estima que ocurre en menos de un caso por cada 10.000-100.000 expuestos. Algunos anestésicos locales usados en Odontología como la epinefrina y ciertos antibióticos como Azitromicina, ciprofloxacina, claritromicina, eritromicina, clindamicina, levofloxacina, moxifloxacina, ofloxacina, trimetroprim- sulfametoxazol pueden provocar un síndrome de Q-T largo. Estos pacientes con el Síndrome de Q-T Largo tienen más riesgo de sufrir endocarditis bacteriana, por lo que se aconseja como mejor manera preventiva para evitar la endocarditis en estos pacientes, la prevención de la caries.
PALABRAS CLAVE
Cardiopatía congénita, síndrome de Q-T Largo, odontología, interacciones farmacológicas, clínica, antibiótico, anestesia local. INTRODUCCIÓN
Las cardiopatías congénitas no son enfermedades raras, siendo su incidencia del 1% de los recién nacidos vivos.(1) El Síndrome de Q-T largo se define como una anomalía del sistema eléctrico del corazón. (2) Es uno de los síndromes hereditarios más frecuentes asociados a muerte súbita. (3) Se ha denominado una canalopatía, una enfermedad de canales iónicos. Sabemos que el potencial de acción cardiaco depende de un delicado equilibrio entre las corrientes iónicas de despolarización y repolarización; si hay una alteración en los genes que codifican dichos canales, se ve traducida en un desequilibrio de dichas corrientes. La repolarización ventricular normal se produce por el equilibrio entre las corrientes de entrada de sodio y calcio, y la corriente de salida de potasio. Estas últimas son varias pero se pueden agrupar en dos, aquellas encargadas de la repolarización en la fase inicial del potencial de acción, que se denominan "corrientes hacia afuera" y aquellas encargadas del resto de repolarización, que se conocen como corrientes rectificadoras tardías. (3,4) Etiología El síndrome de Q-T largo es causado por una mutación en los genes de los canales iónicos a nivel cardiaco para el potasio, calcio y sodio, dependiente del cromosoma afectado. Así se determinará si se trata de un síndrome de Romano Ward (5 subtipos) Jervel Lange Nielsen (2 subtipos) y dos subtipos denominados síndrome de Timothy (afecta los canales de calcio) y el síndrome de Anderson (afecta a canales de potasio). (5, 6, 7,8)
El intervalo QT es la representación eléctrica de la sístole ventricular, tanto del periodo de despolarización como de repolarización, cuando hay un fallo en algún canal de intercambio de iones, aparece el Síndrome de Q-T largo y sus diferentes subtipos. El síndrome de Q-T largo se clasifica en 8 subtipos, el SQTL subtipo I se presenta en un 45-50% de los casos, el SQTL subtipo II en un 40-45%, el subtipo III en un 5-15%, subtipo IV afecta al 1% de los casos, subtipo V al 1%, subtipo VI al 1% y los subtipos VII y VIII al 0,5% de los casos. (9, 10, 11,12)
Esta alteración puede ser congénita y con frecuencia tener un patrón hereditario o puede ser adquirida principalmente ante el uso de algunos medicamentos. Este síndrome se ha asociado a síncope congénito, arritmias ventriculares e inclusive con muerte súbita. (13,14) Diagnóstico
La bradicardia fetal puede ser una de las primeras manifestaciones clínicas en el síndrome de Q-T largo. Si la sospecha de la enfermedad es muy alta, la amniocentesis a partir de las 16 semanas de gestación, puede ser de utilidad para el diagnóstico, que resulta sencillo cuando alguno de los padres es conocido como portador de una mutación determinada. (15)
Las manifestaciones clínicas pueden variar o las personas mostrarse asintomáticas, puede existir una historia de síncope recurrente o bien que la muerte súbita será la primera manifestación de la enfermedad. Comúnmente, las manifestaciones clínicas consisten en episodios sincopales, habitualmente provocados por ansiedad, dolor, ruidos bruscos o ejercicio. Puede haber pródromos como palpitaciones, náuseas, vómitos, cianosis, malestar general y cefalea. (16)
Existen ciertos fármacos que pueden prolongar el intervalo QT, en concreto la taquicardia helicoidal secundaria a fármacos no antiarrítmicos es un evento raro; se estima que ocurre en menos de un caso por cada 10.000-100.000 expuestos. Figura. 1: Fármacos que prolongan el intervalo QT.
Fármacos de aplicación cardiológica
Dopamina, amiodaron, Sotalol, dobutamina, flecainida, ibutilide, efedrina, norepinefrina, quinidan, amirinona, mirinona. Fármacos de aplicación psiquiátrica
Amiptrilina, hidrato de cloral, citalopram, cloropromazina, doxepina, droperidol, fluoxetina, haloperidol, imipramina, litio, metadona, metifenidrato, paroxetina, otanzapina, risperidona, tioridazin, ketanserina. Fármacos de aplicación gastroenterológica
Cisaprida, domperidona, octreótida, ondansetrón, sibutramida, droperidol. Fármacos de aplicación neumológica
Salbutamol, salmeterol, Terbutalina. Fármacos antimicrobianos
Azitromicina, ciprofloxacina, claritromicina, eritromicina, clindamicina, levofloxacina, moxifloxacina, ofloxacina, trimetroprim- sulfametoxazol. Fármacos anestésicos
Epinefrina. Fármacos antifúngicos
Fluconazol, itraconazol, ketoconazol, voriconazol. (13, 17,18) Tratamiento odontológico
Las lesiones endoteliales del corazón o los vasos, ocasionan trombos locales que pueden infectarse a partir de bacterias u hongos del torrente sanguíneo (endocarditis, endarteritis, flebitis). Las endocarditis en niños sin cardiopatía ni otros factores de riesgo es poco frecuente, en menos de un 10% de los casos. La base teórica de la profilaxis de la endocarditis puede resumirse así: cuando se lesionan y sangran los endotelios o epitelios colonizados por bacterias saprófitas, éstas pueden pasar al torrente sanguíneo dando lugar a pequeñas bacteriemias, que podrían infectar con mayor facilidad un endocardio dañado que uno sano. Clásicamente se ha recomendado profilaxis antibiótica ante procedimientos dentales o quirúrgicos con riesgo de bacteriemia en todas las cardiopatías menos en la comunicación interauricular tipo ostium secundum. También se aconseja la profilaxis en los 6 meses posteriores a la cirugía o colocación de dispositivos de cierre de cateterismo, período en el cual se supone que el material de sutura o cierre se habrá epitelizado. Sin embargo, aunque muchos autores recomiendan la profilaxis de antibiótico previo a procedimientos dentales en estos pacientes; el Instituto Nacional para la Salud y Excelencia Clínica del Reino Unido en 2008, desaconsejó la profilaxis antibiótica para estos pacientes, recomendando que la mejor manera de prevenir la endocarditis sea la prevención primaria de la caries. (19,20) OBJETIVO GENERAL Conocer las características clínicas del Síndrome de Q-T largo y poder tratarlos en la consulta de Odontología. MATERIAL Y MÉTODOS
Se ha hecho revisión bibliográfica en PubMed, Scopus; insertando palabras clave como "Cardiopatía congénita, síndrome de Q-T Largo, odontología, interacciones farmacológicas, clínica, antibiótico, anestesia local". Se seleccionaron artículos científicos de revisiones sistemáticas y casos clínicos.
A propósito del tema, desarrollamos un caso clínico del tema que nos ocupa. RESULTADOS
Caso clínico
Paciente de 5 años de edad que acude a la consulta de Odontología del Centro de Salud por caries en dentición temporal. El paciente desde pequeño, fue diagnosticado con el Síndrome de Q-T Largo Tipo V, estando en tratamiento desde los 4 años con tratamiento de propanolol 6 mgr (1mgr/1ml) cada 8 horas.
El paciente fue diagnosticado del Síndrome de Q-T largo mediante un electocardiograma en los primeros meses de vida, revisándolo el Especialista en Cardiología a la edad de 4 meses, 9-12 meses, 2 años, 3 años y 4 años; informando el Cardiólogo que es poco colaborador y no es autosuficiente en las comidas y que presenta relajación de esfínteres de manera nocturna. Conocemos también que la madre también padece el Síndrome de Q-T Largo.
Preguntamos al pequeño sobre sus hábitos de cepillado de dientes, a lo que responde que se cepilla cuando se acuerda, si acaso, una vez al día los dientes. Tras la exploración bucodental, apreciamos caries en dentición temporal en los molares temporales 74-75-84-85 de pequeño extensión, que puede involucrar únicamente la afectación amelodentinaria. 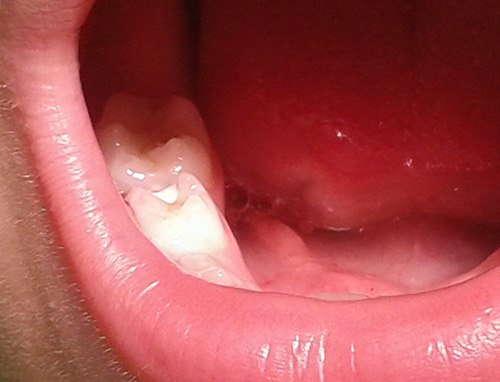 Figura.1: Paciente de 5 años con Síndrome de QT-Largo Tipo V, con caries en 74-75-85-84, en un niño no colaborador. Aconsejo a la madre que acude con su hijo para tratar de manera conservadora esos molares temporales, para evitar así la propagación de infección bacteriana al torrente circulatorio. Doy instrucciones de que la anestesia más segura para utilizar en este tipo de pacientes, es la anestesia tipo amida, sin vasonconstrictor; y se aconseja la administración de una profilaxis antibiótica de penicilina, 50 mgr/kg/peso una hora antes de la anestesia y del tratamiento conservador de esos molares temporales. DISCUSIÓN
El Síndrome Q-T Largo es una canalopatía que puede comprometer la vida, ya que puede presentar una mayor probabilidad de presentar un evento mayor; como síncope, paro cardíaco y muerte súbita. Según Priori et al, en 647 pacientes tratados, mostraron que la probabilidad de presentar un evento mayor, como síncope, paro cardíaco, muerte súbita; antes de los 40 años es alto, mayor del 50%, cuando el QTc es mayor de 500msg en el Síndrome de Q-T Largo 1, en el Síndrome de Q-T Largo 2 o bien en el sexo masculino cuando existe Síndrome de Q-T Largo 3. (21)
Según Medeiros (22) debe considerarse un alto riesgo de padecer el Síndrome Q-T Largo asociado a sordera congénita, síncope recurrente por taquiarritmias ventrículares malignas, bloqueo aurícula-ventriculares y mujeres en el posparto.
Es por tanto de destacar que algunas arritmias, como miocardiopatía hipertrófica y el síndrome de Q-T Largo sin sordera son autonómicos dominantes. (23) Según Jenkins (24) si la madre o alguno de sus hijos tienen una cardiopatía congénita de gravedad, es conveniente que se realice un control ecocardiográfico fetal entre las semanas 16 y 20 para comprobar si el niño tiene Síndrome de Q-T Largo. CONCLUSIONES
Este Síndrome de Q-T Largo se ha asociado a síncope cardiogénico, arritmias ventriculares e inclusive muerte súbita. Dado el incremento en el reconocimiento de esta patología y sus riesgos, los odontólogos deben conocer las posibles etiopatogenias de ciertos antibióticos y anéstesicos que pueden prolongar el intervalo QT, y originar una crisis en este tipo de pacientes. BIBLIOGRAFÍA 1. Arias López I, Martínez Tallo E, Campo Sanpedro F, Cardesa García JJ. Incidencia de las cardiopatías congénitas en la provincia de Badajoz. An Pediatr ( Barc) 2008;69:23-7.
2. Svedjeholm R, Dahlin L, Lundberg C, Szabo Z, Kagedal B, Nylander E. Are electrocardiographic Q-wave criteria reliable for diagnosis of perioperative myocardial infarction after coronary surgery?. Eur J Cardiothorac Surg 1998;13(6):655-61.
3. Iturralde-Torres P, Medeiros-Domingo A. Genética en los síndromes de QT prolongado. Arch Cardiol Mex 2009;79 (2 Suppl):26-30.
4. Monteforte N, Napolitano C, Priori SG. Genética y arritmias: aplicaciones diagnósticas y pronósticas. Rev Esp Cardiol 2012;65:278-86.
5. Chiang CE, Roden DM. The long QT syndromes: genetic Basic and clinical implications. J Am Coll Cardiol 2000 Jul;36(1):1-12.
6. Gutiérrez O, Araya V. Manual de arritmias cardiacas: Guía diagnóstica y terapeútica. Ed. Universidad de Costa Rica, 2002.
7. Napolitano C, Priori SG, Schwartz PJ. Genetic testing in the Long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice. JAMA 2005;294:2975-80.
8. Sovari A, Cocheril A, Assadi R, Zareba W, Rosero S. Long QT Syndrome. January 3, 2007. E-medicine.
9. Montero Chacón LB, Quesada Ramírez MA. Síndrome de QT Largo: Revisión bibliográfica. Rev Médica de Costa Rica y Centroamérica LXVI 2009;587:105-10.
10. Schwartz PJ, Priori SG, Spazzolini C. Genotype-phenotype correlation in the long-QT syndrome gene-specific triggers for life-threatening arrhythmias. Circulation 2001;103:89-95.
11. Beaufort-Krol GC, van der Berg MP, Wilde AA. Developmental aspects of long QT syndrome type 3 and Brugada syndrome on the basis of a single SCN5A mutation in childhood. J Am Coll Cardiol 2005;46:331-7.
12. Mohler PJ, Bennett V. Ankryin-based cardiac arrhythmias: a new class of channelopathies due to loss of cellular targeting. Curr Opin Cardiol 2005;20:189-93.
13. Arizona Center for Education and Research on Therapeutics the Critical Path Institute. [Internet] Revisado 2008.
14. Berul C. Congenital Long-QT Syndromes. Who`s at risk for Sudden Cardiac Death? Circulation 2008;117:2178-80.
15. Tester DJ, McComack J, Ackerman MJ. Prenatal molecular genetic diagnosis of congenital long QT syndrome by strategic genotyping. Am J Cardiol 2004;93:788-91.
16. Gómez-Gómez M, Danglot-Banck, Santamaría-Díaz H. Long QT syndrome in children. Rev Mex Pediatr 2008;75(3):121-31.
17. Tong KL, Lau YS, Teo WS. Case series of Drug-Induced Long QT Syndrome and Torsade de Pointes. Singapore Med J 2001;42(12):566-70.
18. Fitzgerald PT, Ackerman MJ. Drug-induced torsades de pointes: the evolving role of pharmacogenetics. Heart Rhythm 2005;2:S30-7.
19. Vitoria Miñana I, De Baturelli Castillo A. Promoción de la salud bucodental. En: Recomendaciones PrevInfad/PAPPS. [Internet] 2009
20. Smith P. Primary Care in children with congenital Herat disease. J Pediatr Nurs 2001;16:308-19.
21. Priori SG, Schwartz PJ, Napolitano C. Risk stratification in the long-QT syndrome. N Engl J Med 2003;348:1866-74.
22. Medeiros Domingo A, Hernández Cruz A, Iturralde P, Tusié-Luna T. Nuevas perspectivas en el síndrome de QT largo. Rev Invest Clin 2007;59(1):57-72.
23. Moreno García M, Gómez Rodríguez MJ, Barreiro Miranda E. Genética de las cardiopatías congénitas. An Esp Pediatr 2000;53:30-9.
24. Jenkins KJ, Correa A, Feinstein JA. Nonin-herited risk factors and congenital cardiovascular defects: current knowledge. A scientific statement from the American Heart Association council on cardiovascular disease in the young. Endorsed by American Academy of Pediatrics. Circulation 2007;115:2995-3014.
|